July 29, 2015, 12:29 a.m. by Rosalind Team
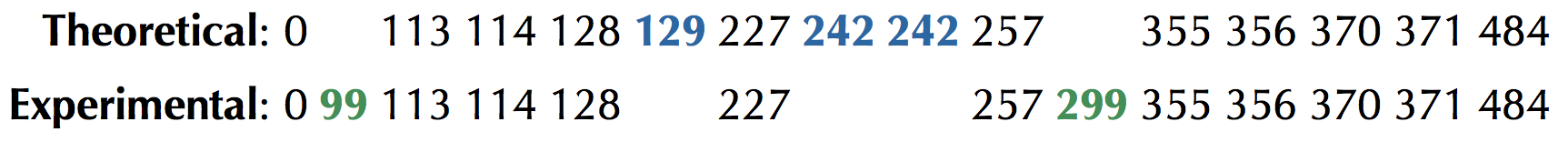
We have thus far worked with theoretical spectra of cyclic peptides, in which the mass of every subpeptide is given. This inflexibility presents a practical barrier, since mass spectrometers generate spectra that are far from ideal — they are characterized by having both false masses and missing masses. A false mass is present in the experimental spectrum but absent from the theoretical spectrum; a missing mass is present in the theoretical spectrum but absent from the experimental spectrum (see Figure 1).
To generalize “Find a Cyclic Peptide with Theoretical Spectrum Matching an Ideal Spectrum” to handle “noisy” spectra having false and missing masses, we need to relax the requirement that a candidate peptide’s theoretical spectrum must match the experimental spectrum exactly, and instead incorporate a scoring function that will select the peptide whose theoretical spectrum matches the given experimental spectrum the most closely. Given a cyclic peptide Peptide and a spectrum Spectrum, we define Score(Peptide, Spectrum) as the number of masses shared between Cyclospectrum(Peptide) and Spectrum. Recalling Figure 1, if
then Score("NQEL", Spectrum) = 11.
To limit the number of candidate peptides under consideration, we will use a Leaderboard, which holds the N highest scoring candidate peptides for further extension. At each step, we will expand all candidate peptides found in Leaderboard by adding every possible amino acid to the end. Then, we will eliminate those peptides whose newly calculated scores are not high enough to keep them on the Leaderboard. This idea is similar to the notion of a “cut” in a golf tournament; after the cut, only the top N golfers are allowed to play in the next round, since they are the only players who have a reasonable chance of winning.
To be fair, a cut should include anyone who is tied with the Nth-place competitor. Thus, Leaderboard should be trimmed down to the “N highest-scoring peptides including ties”, which may include more than N peptides. Given a list of peptides Leaderboard, a spectrum Spectrum, and an integer N, Cut(Leaderboard, Spectrum, N) returns the top N highest-scoring peptides in <Leaderboard (including ties) with respect to Spectrum. We now introduce LEADERBOARDCYCLOPEPTIDESEQUENCING. In what follows, the 0-peptide is the peptide "" containing no amino acids.
Given: An integer N and a collection of integers Spectrum.
Return: LeaderPeptide after running LeaderboardCyclopeptideSequencing(Spectrum, N).
10 0 71 113 129 147 200 218 260 313 331 347 389 460
113-147-71-129
Note
The peptide scoring function in LEADERBOARDCYCLOPEPTIDESEQUENCING currently assumes that peptides are circular, but peptides on the leaderboard should technically be scored as linear peptides until the final step. How would you score a linear peptide against a spectrum?